






 Benefits of a Medical Devices Management System include:
Benefits of a Medical Devices Management System include:
There are a number of benefits of ISO 13485 medical devices management system including:
- Increased access to more markets worldwide with certification
- Meet regulatory requirements and customer expectations
- Increase efficiency, cut costs and monitor supply chain performance
- Outline how to review and improve processes across your organisation
- Demonstrate that you produce safer and more effective medical devices
EN ISO 13485:2012 is a quality management system for medical devices specifically for regulatory purposes. ISO 13485 is an ISO standard that was published in 2003 and it represents the requirements for a quality management system in the design and manufacture of medical devices. The standard superseded two earlier standards that governed medical devices which include ISO 13485:1996 and ISO 13488:1996. The ISO 13485 standard has a supporting guidance document – ISO 14969:2004 – Quality management systems – Guidance on the application of ISO 13485:2003. The guidance document was developed to help medical device designers and manufacturers apply the standard. ISO 13485 is intended for any organisation involved in the design, production, installation, and servicing of medical devices.
While ISO 13485 is a stand-alone document it is generally harmonised with ISO 9001. There is a fundamental difference between the two standards in that ISO 13485 only requires that a company demonstrates that the quality system is implemented and maintained while ISO 9001 requires the organisation to demonstrate continual improvement.
In order to comply with one or more of the European regulatory requirements, complying with ISO 13485 is often seen as the first step. The European directives for medical devices that organisations have to comply with include (90/385/EEC, 93/42/EEC and 98/79/EC) in order to attach CE marking to their products and for other parties that are involved in the process whilst other Directives might also require a CE Marking.
The medical device sector is highly regulated and products in the medical device sector require a CE mark so that they can be sold on the European market. Registration to ISO 13485 demonstrates to regulators and customers that the company is committed to quality and excellence. Your companies quality management system will be continually evaluated by undergoing the regular assessments associated with ISO 13485. This will give your company a competitive edge in domestic and international markets and it will also help your company improve its overall performance.
ISO 13485 is compatible with other “non quality” management systems such as OHSAS 18001 and ISO 14001. This standard is applied to any organisation that produces Class I, II, or III medical devices but does not cover drug manufacturers.
The standard is broken down into 8 main clauses. The first three clauses include scope, normative references and terms and definitions. These are general information for manufacturers about the standard and are not auditable. The five key auditable sections include:
Clause 4 – Quality Management System
This section gives the general requirements. The general requirements include identifying the specific processes and how they interact as well as responsibility for processes that are outsourced.
Clause 5 – Management Responsibility
This section requires management involvement at the level of the person who makes policy and final decisions. A management representative must be appointed by top management.
Clause 6 – Resource Management
This section contains requirements for provision of resources. The management have to ensure there are adequate facilities including tools, space and equipment including computer systems.
Clause 7 – Product Realisation
This is the section of the standard that most effects what people in the company do daily. This section covers everything that is needed to realise a product including customer requirements, designing and manufacturing, installing and supporting a medical device.
Clause 8 – Measurement, Analysis and Improvement
This section provides feedback and other information that enables management to maintain the effectiveness of the quality management system.
Requirements of the standard may only be excluded if they do not absolve of responsibility to meet the requirements and if they do not affect ability to provide conforming product. Permissible exclusions are limited to product realisation (clause 7) and must be justified with details in the quality manual. Otherwise conformity should not be claimed.
When making medical devices there is a risk to a person’s life therefore ISO 13485 requires that organisations establish documented requirements for risk management in the product realisation process. Risk management includes risk assessment, risk analysis and risk reduction. Risk Assessment is identifying risks. Risk Analysis is looking at the probability and severity of all hazardous situations. Risk reduction is reduction, mitigation (labelling), elimination of risk as much as possible.
Risk management applies to quality management system processes and most importantly it applies to the design of the device, manufacturing and supporting services. Risk management is such an important process that ISO 13485 requires risk management is done in accordance with ISO 14971. The international standard for medical device risk assessment is ISO 14971.
These 8 principles below are not auditable but they are fundamental attributes of any quality management system. The principles have been taken from ISO 9000:2005 Quality Management Systems – Fundamentals and Vocabulary and have served as a basis for the ISO 9001:2008 standard. The 8 principles include:
- Customer focus
- Leadership
- Involvement of people
- Process approach
- System approach to management
- Continual improvement
- Factual approach to decision making
- Mutually beneficial supplier relationships
PDCA model applied to ISO 13485 Quality Management System
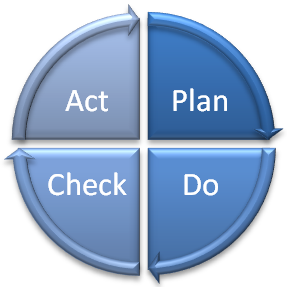 The Plan – Do – Check – Act (PDCA) cycle is the foundation of all ISO management system standards. The cycle ensures development, continuous improvement and control of the management system in question.
The Plan – Do – Check – Act (PDCA) cycle is the foundation of all ISO management system standards. The cycle ensures development, continuous improvement and control of the management system in question.
The PDCA cycle ensures constant monitoring of your organisation’s effectiveness. It consists of the following:
- Plan – establishing the architecture of your quality management system is covered in clause 4.1 of the standard where it requires the identification of the processes, their success criteria, the inter-relationship between processes and the system for checking your results
- Do – implementing the plans and using the quality management system
- Check – reviewing whether the results are satisfactory at appropriate intervals against the ISO 9001 requirements
- Act – improving the quality management system or acting on the challenges and issues found in the reviews
An organisation will probably already have an effective quality management system but it is most likely informal and not well documented. A more systematic approach to achieving organisation’s objectives is provided by ISO 9001. This should not result in excessive bureaucracy or paperwork and lack of flexibility. It should not be a financial burden either. The quality management systems should be viewed as an investment and the return on the investment is the previously mentioned benefits.
Plan
The PDCA cycle starts with management because they identify appropriate processes and relevant areas of focus.
Process Identification:
An essential requirement for a practical system is an appropriate process and the key is starting with two processes which are Management and Operations. Next decide if sub-processes are required instead of working “bottom up”. An owner is required for every process who is responsible for the activities that relate to the success criteria of the process.
Planning and Review:
A quality manual and a number of documents outlining procedures are required before implementation in order to successfully plan your quality system. The areas of documentation are:
- Document control
- Records control
- Internal audits
- Non – conforming product
- Corrective action
- Preventative action
Fundamental direction:
The fundamental direction of the QMS should be established by owners or managers of your organisation using the quality policy. When designing the Quality Policy there are several aspects that have to be thought through such as:
- Strategy – should follow from the Quality Policy and the business environment
- Process criteria – should be aligned to the strategy
- Customer focus – system processes have to be designed to ensure customer satisfaction
- Resources – human, technological and environmental resources have to be put in place. The QMS requires that each company establish a way that their staff are competent
Do
The system has to be used to see that it works the way it was intended to. It will be necessary to use the procedures, forms, equipment and instructions in the way it was planned. This part of the process should be fairly easy to implement with the direction from your management and the assigned resources. It is important that the processes all along the supply chain should be planned and defined. This might include:
- Sales
- Purchasing
- Research and development
- Manufacturing
- Delivery
Some of the steps might not apply to your organisation as ISO 9001 certification is designed for every type of organisation .
Check
The results of the QMS should be reviewed at appropriate intervals. When the system is new the intervals will be short but can be longer once the QMS becomes mature. The reporting of results against the process success criteria should be done regularly and then be used by management to ensure that the business is on track. The records should be appropriately designed to facilitate prompt recording as well as the early detection of problems.
The management review is a key milestone in evaluating the QMS and it is a meeting which assesses whether the QMS has succeeded in meeting:
- Strategic objectives
- Process success criteria
- ISO 9001 requirements
A key metric that has to be reviewed is perceived customer satisfaction. Handling complaints is not enough as customers could just move their businesses to a competitor. Internal audits are probably the most important characteristic of a successful quality management system. If an organisation does not carry out internal audits it is likely that the organisation will have their certification revoked as their system is probably out of control.
Act
Corrective action or preferably preventative action can be used to tackle challenges. Corrective actions must be recorded and preventative actions should be designed for recurring problems. The following questions should be asked as a checklist:
- Customer focus – Have you found out what the customer’s current and future need and expectations are at a strategic level?
- Quality policy – Does it really suit your organisation and reflect your customer’s expectations, your vision and mission – and the requirements of the standard?
- Objectives – Are all the objectives measurable and linked to both the processes and to the strategies?
- Plan the system – Have all the responsibilities been identified and communicated? Does everyone know what they need to do to contribute to the success of the business – and the QMS?
- Review at regular intervals – Are the results of the QMS being reviewed and compared against planned results? Is action being taken to improve areas where results are not quite as good as planned?
- Principles – management should review the 8 principles mentioned earlier and how well the system delivers against these.
In order to get certified for ISO 13485 Medical Devices Quality Management System there are a number of steps involved:
Applying
The first step involves the application for certification
Gap Assessment
This is where:
- An on-site analysis of your current system is conducted
- This will then be assessed against the standard
- A report is then prepared to highlight the gaps between your current system and the standard
The gap assessment is optional and is not required for the certification process.
Preliminary Assessment – Stage 1
Your documentation has to be inspected and various areas have to be reviewed including:
- The proposed scope of your registration
- The status of implementation of your management system
- The appropriate regulatory and legal requirements
- Your management policies and objectives
- Whether the system addresses the key areas of your business
- Your site-specific activities – top level process review
- Your key management elements, e.g. internal audits, reviews and complaints procedures
- Your readiness to move onto Stage 2 of the assessment, the Registration Assessment
There should be a period of several weeks between preliminary assessment and registration assessment so that any issues can be sorted in relation to the preliminary assessment. If there are major non-conformities a second preliminary assessment will have to be carried out.
Registration Assessment – Stage 2This involves a full review of your management system in order to confirm that your management system is controlled. When the registration assessment is complete a notified body issues a detailed report along with the outcome which recommends registration or not. If any issues arise during the assessment you will be expected to submit an action plan which should describe what changes are to be made to the management system in order to reduce or eliminate the risk of the same issues occurring again.
Surveillance and Re-assessment
A notified body visits each company at least once a year to make sure the management system is being maintained and that it is achieving it’s expected outcomes. A part of the management system is reviewed in depth during each visit.
There is an expiry date on the certificate and the certificate expires every three years. Before the expiry date occurs a detailed assessment of the whole management system is undertaken to ensure every element of the system is performing satisfactorily and the results of the previous visits are taken into account.
During the registration period, changes are inevitable. In order to make sure the management system remains sound the notified body works with each registered organisation. Usually, change can be reviewed and assessed during routine surveillance visits. The notified body reserves the right to suspend or revoke certification in cases where change leads to the breakdown of the system.
